



Hemofili A är mest typiskt en genetisk blödningsstörning orsakad av ett saknat eller defekt koagulationsprotein som kallas faktor VIII. Det kallas också klassisk hemofili eller faktor VIII-brist. I sällsynta fall är det inte ärvt, men i stället orsakat av en onormal immunreaktion i kroppen.
Personer med hemofili A blöder och blåmärken lätt, och deras blod tar lång tid att bilda blodproppar. Hemofili A är ett sällsynt, allvarligt tillstånd som inte har botemedel, men är behandlingsbart.
Läs vidare för att få en bättre förståelse för denna blödningsstörning, inklusive orsaker, riskfaktorer, symtom och eventuella komplikationer.
Vad orsakar hemofili A?
Hemofili A är oftast en genetisk störning. Detta innebär att det orsakas av förändringar (mutationer) till en viss gen. När denna mutation är ärvad, går den ner från föräldrar till barn.
Den specifika genmutation som orsakar hemofili A leder till en brist i en koagulationsfaktor som kallas faktor VIII. Din kropp använder en mängd olika koagulationsfaktorer för att hjälpa till att bilda blodproppar vid ett sår eller skada.
En blodpropp är en geliknande substans gjord av element i kroppen som kallas blodplättar och fibrin. Clots hjälper till att stoppa blödningen från en skada eller skära och låta det läka. Utan tillräckligt med faktor VIII kommer blödningen att förlängas.
Mindre ofta uppträder hemofili A slumpmässigt hos en person utan tidigare familjehistoria av sjukdomen. Detta är känt som förvärvat hemofili A. Det är typiskt orsakat av en persons immunsystem som felaktigt gör antikroppar som attackerar faktor VIII. Förvärvat hemofili är vanligare hos personer mellan 60 och 80 år och hos gravida kvinnor. Förvärvat hemofili har varit känt att lösa, till skillnad från den ärftliga formen.
Hur skiljer sig hemofili A från B och C?
Det finns tre typer hemofili: A, B (även känd som julsjukdom) och C.
Hemofili A och B har mycket liknande symtom men orsakas av olika genmutationer. Hemofili A orsakas av en brist i koagulationsfaktor VIII. Hemofili B är resultatet av en brist i faktor IX.
Å andra sidan beror hemofili C på en faktor XI-brist. De flesta människor med denna typ av hemofili har inga symtom och ofta blödar inte i leder och muskler. Förlängd blödning sker vanligen endast efter en skada eller operation. Till skillnad från hemofili A och B är hemofili C vanligast hos Ashkenazi-judar och påverkar både män och kvinnor lika.
Faktor VIII och IX är inte de enda koagulationsfaktorer som kroppen behöver för att bilda koaguler. Andra sällsynta blödningsstörningar kan inträffa när det finns brister i faktorerna I, II, V, VII, X, XII eller XIII. Brister i dessa andra koagulationsfaktorer är emellertid extremt sällsynta, så det är inte mycket känt om dessa störningar.
Alla tre typer av hemofili anses vara sällsynta sjukdomar, men hemofili A är den vanligaste av de tre.
Vem är i riskzonen?
Hemofili är sällsynt - det förekommer hos endast 1 av 5 000 födda. Hemofili A förekommer lika i alla ras och etniska grupper.
Det kallas ett X-länkat tillstånd eftersom mutationen som orsakar hemofili A finns på X-kromosomen. Män bestämmer barnets könskromosomer, vilket ger en X-kromosom till döttrar och en Y-kromosom till söner. Så honor är XX och män är XY.
När en far har hemofili A, ligger den på sin X-kromosom. Om man antar att moderen inte är bärare av eller har störningen, kommer ingen av hans söner att arva villkoret, eftersom alla hans söner kommer att ha en Y-kromosom från honom. Men alla hans döttrar kommer att vara bärare eftersom de fick en hemofili-påverkad X-kromosom från honom och en opåverkad X-kromosom från moderen.
Kvinnor som är bärare har 50 procent chans att överföra mutationen till sina barn, eftersom en X-kromosom påverkas och den andra inte är. Om hennes söner ärva den drabbade X-kromosomen kommer de att få sjukdomen, eftersom deras enda X-kromosom är från deras mamma. Alla döttrar som ärver den drabbade genen från sin mamma kommer att vara bärare.
Det enda sättet en kvinna kan utveckla hemofili är om fadern har hemofili och mamman är en bärare eller har sjukdomen också. En kvinna kräver hemofili-mutationen på båda X-kromosomerna för att visa tecken på tillståndet.
Vad är symtom på hemofili A?
Personer med hemofili A blöder oftare och under längre tid än människor utan sjukdom. Blödningen kan vara intern, såsom inom leder eller muskler, eller externt och synligt, från nedskärningar. Blödans allvar beror på hur mycket faktor VIII en person har i sin blodplasma. Det finns tre nivåer av svårighetsgrad:
Allvarlig hemofili
Cirka 60 procent av personer med hemofili A har allvarliga symptom. Symtom på svår hemofili är:
- blödning efter en skada
- spontan blödning
- täta, svullna eller smärtsamma leder som orsakas av blödning i lederna
- näsblod
- kraftig blödning från en mindre skära
- blod i urinen
- blod i avföringen
- stora blåmärken
- blödande tandkött
Måttlig hemofili
Ungefär 15 procent av personer med hemofili A har ett måttligt fall. Symtomen på måttlig hemofili A liknar allvarlig hemofili A, men är mindre allvarliga och uppträder mindre ofta. Symtom inkluderar:
- långvarig blödning efter skador
- spontan blödning utan uppenbar orsak
- blåmärken lätt
- gemensam styvhet eller smärta
Mild hemofili
Omkring 25 procent av hemofili A-fall anses vara milda. Ofta görs en diagnos först efter en allvarlig skada eller en operation. Symtom inkluderar:
- långvarig blödning efter allvarlig skada, trauma eller kirurgi, såsom en tanduttag
- lätt blåmärken och blödning
- ovanlig blödning
Hur diagnostiseras hemofili A?
En läkare gör en diagnos genom att mäta nivån av faktor VIII-aktivitet i ett blodprov.
Om det finns en familjehistoria för hemofili, eller om en mamma är en känd bärare, kan diagnostiska tester göras under graviditeten. Detta kallas prenatal diagnos.
Vad är komplikationerna av hemofili A?
Repeterande och överdriven blödning kan leda till komplikationer, särskilt om det går obehandlat. Dessa inkluderar:
- allvarlig anemi
- gemensam skada
- djup inre blödning
- neurologiska symptom från blödning i hjärnan
- en immunreaktion mot koagulationsfaktorbehandling
Vid infusioner av donerat blod ökar också risken för infektioner, såsom hepatit. Men nuförtiden doneras blod noggrant före transfusion.
Hur behandlas hemofili A?
Det finns ingen botemedel mot hemofili A och de med sjukdomen kräver livslång behandling. Det rekommenderas att individer får behandling vid ett specialiserat hemofilibehandlingscenter (HTC) när det är möjligt. Förutom behandling ger HTCs resurser och stöd.
Behandling innebär att man ersätter den saknade koagulationsfaktorn via transfusioner. Faktor VIII kan erhållas från bloddonationer, men är nu vanligen skapad konstgjort i ett labb. Detta kallas rekombinant faktor VIII.
Behandlingsfrekvensen beror på svårighetsgraden av sjukdomen:
Mild hemofili A
De med lätta former av hemofili A kan bara behöva ersättningsbehandling efter en blödningsepisod. Detta kallas episodisk eller på begäran behandling. Infusioner av ett hormon som kallas desmopressin (DDAVP) kan bidra till att stimulera kroppen att frigöra mer koagulationsfaktor för att stoppa blödningsepisoden. Läkemedel som kallas fibrintätningsmedel kan också appliceras på ett sårläge för att främja läkande.
Svår hemofili A
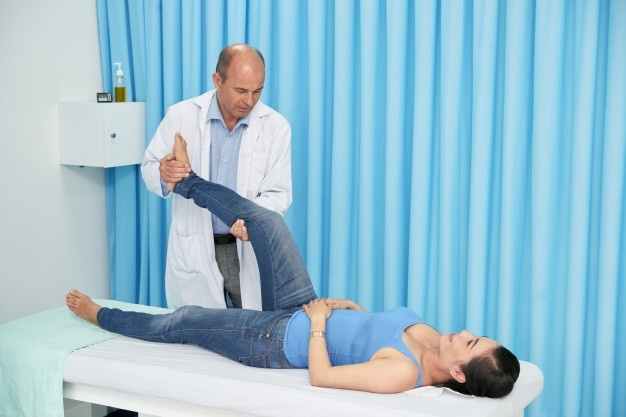
Personer med svår hemofili A kan få periodiska infusioner av faktor VIII för att förhindra blödningsepisoder och komplikationer. Detta kallas profylaktisk terapi. Dessa patienter kan också träna för att ge infusioner hemma. Svåra fall kan behöva fysisk terapi för att lindra smärta orsakad av blödning i lederna. I allvarliga fall är kirurgi nödvändig.
Vad är utsikterna?
Utsikterna beror på huruvida någon får rätt behandling eller ej. Många personer med hemofili A kommer att dö före vuxenlivet om de inte får adekvat vård. Men med rätt behandling förutses en nära normal livslängd.





{kind=link}